8448966
Description
Flashcards by Ashutosh Kumar, updated more than 1 year ago
|
|
Created by Ashutosh Kumar
about 7 years ago
|
|
| Question | Answer |
| Life span of RBCs: | 107 days |
| Reasons for reduced red cell survival in the circulation: | Reduced red cell survival in the circulation: Abnormal RBCs. Increased destruction of RBCs. |
| Marrow responds with increased production of RBCs, resulting in: | Marrow responds with increased production of RBCs, resulting in: Increased reticulocytes, which are large young RBCs. Residual RNA in reticulocytes permits their identification via polychromasia on staining. |
| Main tests for haemolysis: Other tests for haemolysis: | Main tests for haemolysis: 1. Blood screen: haemoglobin and RBC morphology 2. Reticulocyte count. 3. Bilirubin: A metabolic product of haem; unconjugated elevated predominantly. 4. Haptoglobin: binds to haemoglobin and is consumed; goes low even with mild haemolysis. Other tests for haemolysis: LDH (increases with haemolysis but not specific) Heinz bodies Hb electrophoresis- haemoglobinopathies Red cell enzyme studies (G6PD) |
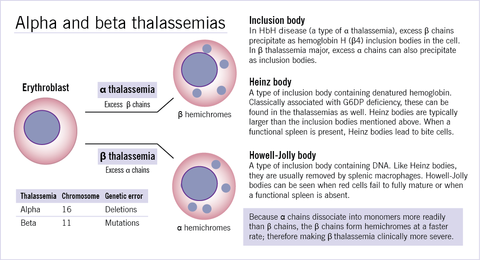
| Inherited haemolytic anaemia categories: | Inherited haemolytic anaemias: Cell membrane of cytoskeletal abnormalities Hereditary spherocytosis Enzymopathies Glucose-6-phosphate dehydrogenase deficiency Haemoglobinopathies Thalassaemia (in alpha thalassaemia excess beta chains cause membrane damage whereas in beta thalassaemia excess alpha chains cause membrane damage) Abnormal globin chain (HbS sickle cell anaemia) |
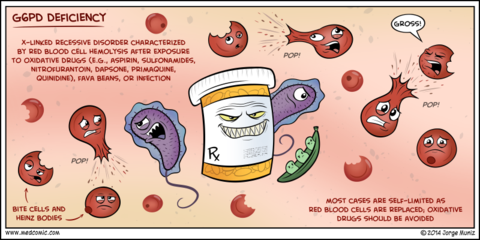
| Glucose-6-phosphate dehydrogenase deficiency: Pathophysiology: Clinical effects: Blood film results: | Glucose-6-phosphate dehydrogenase deficiency: Failure to produce normal amounts of NADPH, which is reduced to NADP to convert GSSG to GSH, which is an important antioxidant called glutathione. As a result Hb and cell membranes become cross linked. Haemolysis occurs because of oxidative damage to Hb and cell membrane. Provides resistance against malaria since the parasite needs NADPH for energy. Features of G6PD: Episodes of Haemolytic anaemia precipitate by illness, drugs and broad beans. Increased reticulocytes Blood film: Reticulocytes and Heinz bodies seen on blood film. They are a blue stained mass of oxidized, denatured and aggregated Hb attached to the inside of the RBC membrane. |
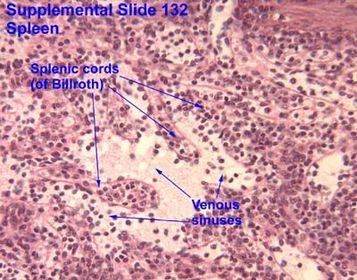
| Spleen the filter: Channel types: | Spleen the filter: A minority (10%) of RBCs have to travel through slow channels (cords) and then transverse into high speed sinuses (S) |
| Thalassaemia: Pathophysiology of haemolysis: | Thalassaemia: Shortened RBC survival as well as impaired haemoglobin production. Beta thalassaemia: Excess alpha chains stick to and damage RBC membranes. Alpha thalassaemia: Excess beta chains precipitate and form HbH bodies. |
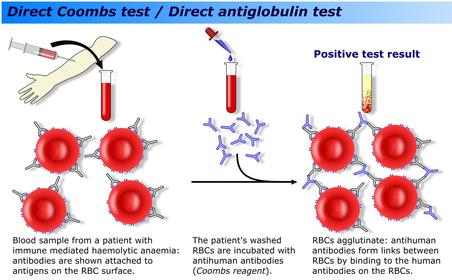
| Autoimmune haemolytic anaemia: Pathophysiology: Test: Treatment options: | Autoimmune haemolytic anaemia: Antibodies are produced against the RBC which then bind to the RBC. The Fc portion of the antibody is recognized by a macrophage which then either: Engulfs the RBC Selectively removes a portion of the RBC membrane resulting in the formation of spherocytes Direct antiglobulin test (DAT)/direct coombs test used to detect AIHA. Treatment options include steroids to suppress immune function and splenectomy (where macrophages primarily come into contact with RBCs). There are consequences to splenectomy; increased susceptibility to developing septicaemia particularly with infection with encapsulated bacteria. Hence 5 yearly vaccinations against Strep. Pneumoniae, Meningococcal and H.Influenza B, wearing a med alert bracelet and early treatment for infections are recommended. |
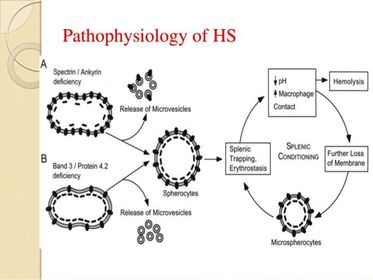
| Hereditary spherocytosis: Pathophysiology: Long term consequences: Treatment: | Hereditary spherocytosis: Due to autosomal dominant mutations of RBC cytoskeletal proteins e.g ankyrin, spectrin, band 3 or protein 4.2 Mutations in ankyrin, spectrin and protein 4.2 results in reduced density of the membrane skeleton, disrupting the stability of the overlying lipid layer releasing band 3 containing microvesicles. Defects of band 3 result in reduced band 3 and its lipid stabilizing effect resulting in microvesicles forming without band 3. In both instances, RBCs form with reduced surface to volume ratio and decreased deformability. As a result, splenic destruction of spherocytes occurs as they pass from the cords to the sinuses by macrophages. Anaemia ensues. Splenectomy may be necessary in some cases. With lifelong haemolytic anaemia, there is increased unconjugated bilirubin, which over time may precipitate forming pigment gallstones. |
| Intravascular haemolysis: Causes: | Intravascular haemolysis: Causes: 1. Mechanical damage: (a) Fibrin strands (thrombotic microangiopathies) Haemolytic uraemic syndrome Thrombotic thrombocytopenic purpura HELLP syndrome (b) Mechanical heart valves 2. Complement induced lysis |
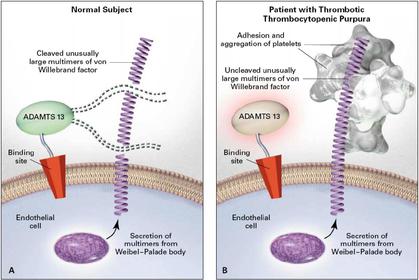
| Thrombotic thrombocytopenic purpura (TTP): Features: Pathophysiology: Mortality: Treatment: | Thrombotic thrombocytopenic purpura (TTP): Features; Neurological, fever, renal impairment, thrombocytopenia and fragmented RBCs. Pathophysiology: Due to a deficiency of ADAMTS13; an enzyme that normally cleaves large multimers of vWF (von willebrand factor). Endothelial cells synthesize vWF as large multimers which project from the endothelial cell into the bloodstream. Such an extensive multimer is cleaved by the ADAMTS13 enzyme into smaller multimers, with only a small multimer jutting out from the endothelial cell. A mutation in ADAMTS13 results in lack of cleavage of the large vWF multimer, and since this is projecting into the bloodstream, platelets become adhere, become activated and aggregate. Untreated mortality is high at 80%. Plasmapheresis reduces this to 20% by essentially using fresh frozen plasma to replace deficient ADAMTS13. |
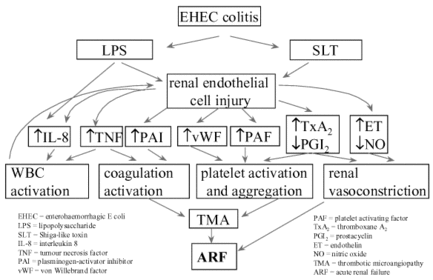
| Haemolytic uraemic syndrome (HUS): Main age group affected: Causative agent: Pathophysiology: Consequences: Treatment: | Haemolytic uraemic syndrome (HUS): Usually occurs in children >90% of cases are due to shiga toxin, produced by organisms such as E.coli 0157 Shiga toxin binds to Gb3 receptor on glomerular endothelium triggering a signalling cascade that either leads to apoptosis or leukocyte binding. The shiga toxin activated endothelial cells become thrombogenic through producing cytokines that activate platelets. In addition, shiga toxin inhibits ADAMTS13 enzyme, resulting in the formation of large multimers of vWF, resulting in platelet adhesion, activation and aggregation. Consequently, a microthrombus forms resulting in microangiopathic haemolysis, where the microthrombus moving through small vessels destroy RBCs, forming schistocytes or fragmented RBCs. Renal failure occurs due to toxicity to the renal endothelium; dialysis required Other organs are usually not involved >90% do not require plasmapheresis |
| HELLP syndrome: Features: Pathophysiology: Consequences: | HELLP syndrome: Haemolysis Elevated Liver enzymes Low Platelets (also RUQ pain, malaise, nausea and vomiting) Caused by maternal-fetal immune imbalance resulting in platelet aggregation, endothelial dysfunction and arterial hypertension. Occurs in pregnancy (>30 weeks). Urgent delivery needed. 10-20% fetal mortality. |
| Heart valve-mechanical haemolysis: Causes: Effect on urine: | Heart valve-mechanical haemolysis: Mechanical valve or paravalvular leak Causes accumulation of iron in renal tubular cells=urinary haemosiderin. |
| Paroxysmal nocturnal haemoglobinuria: Pathophysiology: Patients suffer from: Treatment: | Paroxysmal nocturnal haemoglobinuria: Complement mediated haemolysis Individuals have an acquired deficiency of complement inhibitors on the surface or RBCs. As a result, there is the inability to stop membrane attack complex (MAC) formation on the surface of RBCs resulting in intravascular haemolysis, producing red/orange urine. E.g CD59 is required to stop C8 to C9 step of MAC formation. It is common for mutations to occur in GPI anchored proteins, especially complement inhibitors. In PNH, there is a mutation in PIGA gene in a cell, resulting in subsequent daughter cells through clonal expansion which also have the defect. This mutation means affected cells are unable to synthesis the anchor for GPI proteins, resulting in a deficiency in all GPI proteins. Patients suffer from thrombosis, anaemia (from haemolysis), smooth muscle dystonia (due to NO depletion) and overall impaired quality of life. Treatment: Eculizimab is a monoclonal antibody that binds to C5 and prevents cleavage by C5 convertase to C5a (anaphylatoxin) and C5b (triggers MAC formation). Costs $500,000 NZD. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Want to create your own Flashcards for free with GoConqr? Learn more.