21301037
Description
Mind Map by Ana Cristina Acosta , updated more than 1 year ago
|
|
Created by Ana Cristina Acosta
almost 6 years ago
|
|
Causas Glomerulares
- El ser humano posee en promedio 1.8 millones de capilares glomerulares en forma de ovillo
- Los capilares glomerulares filtran entre 120-180L de plasma al día
- La membrana basal glomerular es una barrera selectiva de tamaño y carga para el paso de
macromoléculas circulantes desde el plasma.
- está controlada por el diámetro del poro, cargas electrostáticas negativas hace que la barrera no
permita el paso de moléculas grandes.
- está controlada por el diámetro del poro, cargas electrostáticas negativas hace que la barrera no
permita el paso de moléculas grandes.
- Excretan no más de 8-10mg de albúmina
diaria
- Puede presentarse casos de proteinuria
benigna/funcional/transitoria
- Ejercicio
- Obesidad
- Embarazo
- Estrés
- Ejercicio
- Puede presentarse casos de proteinuria
benigna/funcional/transitoria
- La membrana basal glomerular es una barrera selectiva de tamaño y carga para el paso de
macromoléculas circulantes desde el plasma.
- Los capilares glomerulares filtran entre 120-180L de plasma al día
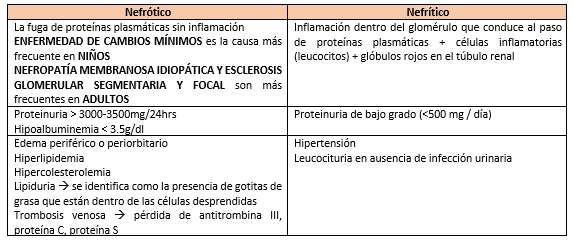
- Las glomerulopatías pueden adoptar manifestaciones clínicas de síndrome nefrótico y de síndrome
nefrítico
- Síndrome
nefrítico
- Edema
Leve
- Hematuria
macroscópica
- oliguria
- Las patologías que se presentan como síndrome nefrítico son
- Glomerulonefritis
postestreptocócica
- Afecta a niños de 2-14 años, sin embargo, en países en
desarrollo es más común en adultos mayores.
Predomina en varones 2:1
- antecedente de infección de
cepas nefritogénicas
- Faringitis
- Impétigo
- Endocarditis
- Faringitis
- fisiopatología se
describe
- - Depósito de inmunocomplejos que contienen
compuestos antigénicos del estreptococo
- - Formación de complejos inmune in situ en el MBG
- - Reactividad
inmune
- - Depósito de inmunocomplejos que contienen
compuestos antigénicos del estreptococo
- histopatología
- hipercelularidad mesangial, endotelial
- infiltrado de
PMN
- depósito inmunitario granulosos en el plano
endotelial de inmunoglobulinas y fracciones del
complemento
- hipercelularidad mesangial, endotelial
- Afecta a niños de 2-14 años, sin embargo, en países en
desarrollo es más común en adultos mayores.
Predomina en varones 2:1
- Nefritis lúpica
- complicación grave del
LES
- presenta más en mujeres
adolescentes de raza negra
- se produce a causa de depósito de complejos
inmunitarios que activan la cascada de
complemento, infiltración leucocítica, activación
de factores procoagulantes y la liberación de
citocinas
- En pacientes con nefritis lúpica se puede observar
hipocomplementemia en el 70-90% de los pacientes y
muchas veces la baja del complemento indica exacerbación
en el cuadro
- complicación grave del
LES
- Nefropatía por IgA
- Lesión más común que causa glomerulonefritis
primaria
- Afecta predominantemente afecta a los varones. Su máxima
frecuencia de presentación se da a los 20-39 años
- fisiopatología
- deposito inicial de IgA en el mesangio
- la inmunoglobulina es de subclase IgA1
- deposito inicial de IgA en el mesangio
- etiología
- Infecciones
- CMV
- H.influenzae
- Toxoplasmosis
- CMV
- Asociaciones genéticas
- Hipersensibilidad a los antígenos alimentarios
- Infecciones
- No existe un tratamiento óptimo, sin embargo, se
recomienda el empleo de IECAS en personas con
proteinurias o deterioro de la función renal
- Lesión más común que causa glomerulonefritis
primaria
- Glomerulonefritis
membranoproliferativa
- Se la conoce también como glomerulonefritis
mesangiocapilar o lobular
- engrosamiento de la membrana basal
glomerular
- engrosamiento de la membrana basal
glomerular
- 70% de los pacientes presentan
hipocomplementemia
- Hay 3 tipos de acuerdo a su Histología
- Tipo I
- suele acompañar a hepatitis C persistente,
enfermedades autoinmunes como él LES
- suele acompañar a hepatitis C persistente,
enfermedades autoinmunes como él LES
- Tipo II y
III
- Idiopáticas
- Idiopáticas
- Tipo I
- Los síntomas iniciales de la GNMP son: proteinuria, hematuria, piuria. Además
síntomas generales como fatiga y malestar
- Se la conoce también como glomerulonefritis
mesangiocapilar o lobular
- Glomerulonefritis
postestreptocócica
- Edema
Leve
- Síndrome
nefrótico
- Edema grave:
anasarca
- Hipocomplementemia
- Las patologías que se presentan como Sindrome Nefótico son:
- Enfermedad de Fabry
- alteración genética ligada al cromosoma
X
- actividad deficiente de la galactosidasa alfa A
- actividad deficiente de la galactosidasa alfa A
- órganos afectados comprenden el endotelio vascular,
corazón, encéfalo y riñones
- forma clásica es que se presente en la
niñez en varones
- glomérulos desarrollan glomeruloesclerosis
de progresión rápida
- La nefropatía por enfermedad de Fabry surge en tercer
decenio
- alteración genética ligada al cromosoma
X
- Síndrome de
Alport
- desarrolla en un 85% de personas con herencia ligada al
cromosoma X
- muestra alteraciones de la cadena de colágenos IV alfa5
- muestra alteraciones de la cadena de colágenos IV alfa5
- Glomeruloesclerosis crónica + sordera
neurosensorial
- desarrolla en un 85% de personas con herencia ligada al
cromosoma X
- Enfermedad de Fabry
- Edema grave:
anasarca
- Síndrome
nefrítico
- Los daños glomerulares pueden
describirse de varias formas:
- las lesiones
histopatológicas
- Proliferativo
- Esclerosante
- Necrotizante
- Proliferativo
- Las alteraciones glomerulares se
presentan por:
- - Disfunción de los
podocitos
- - DM y
amiloidosis
- - Mecanismos
inmunes
- - Disfunción de los
podocitos
- las lesiones
histopatológicas
- Los hallazgos indicativos de daño de origen
glomerular son
- Cilindros
eritrocitarios
- Eritrocitos dismórficos,
acantocitos
- Albuminuria intensa
- Hematuria
- más de 5 eritrocitos por campo
- más de 5 eritrocitos por campo
- Cilindros
eritrocitarios
- La progresión de la enfermedad glomerular
es
- nefritis
intersticial
- fibrosis renal
- Atrofia
tubular
- nefritis
intersticial
Media attachments
{kind=link}
{kind=link}
Want to create your own Mind Maps for free with GoConqr? Learn more.