6526637
Beschreibung
Mindmap von Sofia Berdugo, aktualisiert more than 1 year ago
|
|
Erstellt von Sofia Berdugo
vor mehr als 7 Jahre
|
|
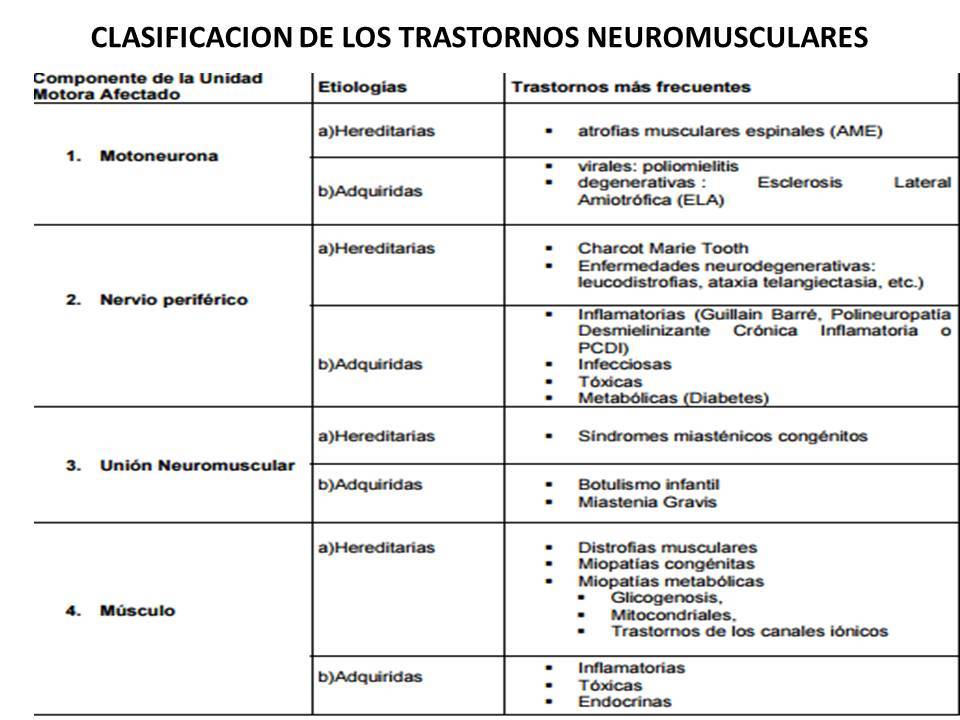
ENFERMEDADES NEUROMUSCULARES
MOTONEURONA 2
- MOTONEURONA
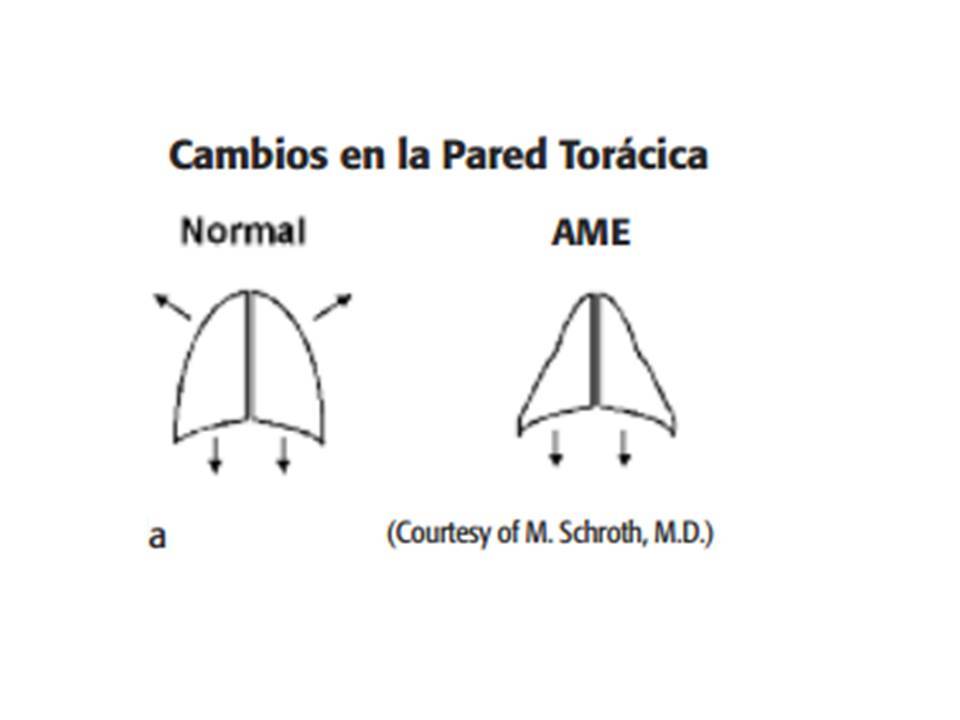
- ATROFIAS ESPINALES AME
- Trastorno hereditario autosómica recesiva de las neuronas motoras
espinales y bulbares .
- que causan atrofia y debilidad musculares, usualmente de forma simétrica y proximal,
con predominio de extremidades inferiores y estando respetada la musculatura facial y
el intelecto
- MANIFESTACIONES
CLINICAS
- Curso progresivo. Predominio en
tronco y cintura pelvica de extremidades
inferiores.
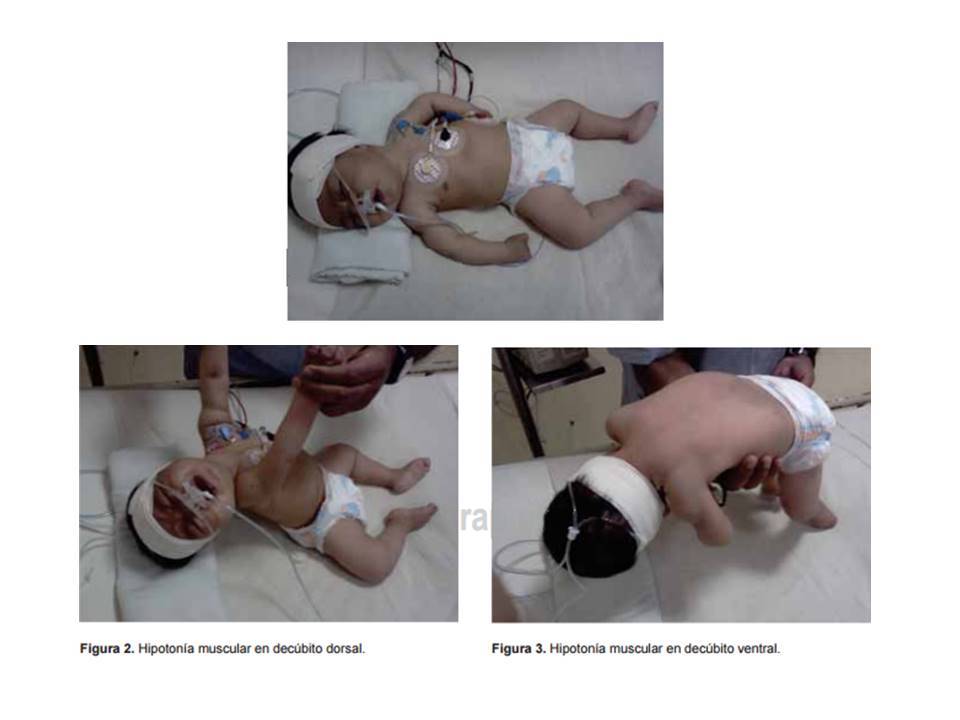
- Hipotonía y debilidad
muscular con abolición de
reflejos osteotendinosos
- Fasciculaciones y tremores
musculares, más visibles en la
lengua (inconstante).
- Curso progresivo. Predominio en
tronco y cintura pelvica de extremidades
inferiores.
- MANIFESTACIONES
CLINICAS
- normalmente ambos padres son
portadores del gen anormal y transmiten
este gen a su hijo
- tipos
- AME Tipo I
- Enfermedad de
Werdnig-Hoffmann.
- DX
- antes de los 6
meses de edad
- escaso control de la cabeza
- rasgo característico los niños no pueden sentarse ni
ponerse de pie SIN asistiencia
- dificultades para la deglución
terminan con gastrostomía
- debilidad en los músculos respiratorios Los pulmones no
se desarrollan completamente
- escoliosis, displasia de
cadera, huesos se vuelven débiles y
pueden romperse con facilidad
- escoliosis, displasia de
cadera, huesos se vuelven débiles y
pueden romperse con facilidad
- debilidad en los músculos respiratorios Los pulmones no
se desarrollan completamente
- Hipotonía de las cuatro
extremidades con posición
característica: brazos extendidos a lo
largo del tronco, pronación de
antebrazos, así como piernas
abiertas y pegadas al plano de apoyo
- dificultades para la deglución
terminan con gastrostomía
- rasgo característico los niños no pueden sentarse ni
ponerse de pie SIN asistiencia
- escaso control de la cabeza
- antes de los 6
meses de edad
- DX
- Enfermedad de
Werdnig-Hoffmann.
- AME Tipo II
- dx
- diagnóstico casi siempre
se realiza antes de los 2
años
- retrasos en los hitos del desarrollo motor
- rasgo distintivo es la capacidad de
mantener una posición sedente sin
soporte
- pueden ponerse de pie con asistencia y el
uso de soportes ortopédicos, pero no
pueden caminar y requieren una silla de
ruedas para movilizarse.
- debilidad músculos
respiratorios dificultad para
toser
- escoliosis, displasia de
cadera, huesos se vuelven
débiles y pueden romperse
con facilidad
- escoliosis, displasia de
cadera, huesos se vuelven
débiles y pueden romperse
con facilidad
- debilidad músculos
respiratorios dificultad para
toser
- pueden ponerse de pie con asistencia y el
uso de soportes ortopédicos, pero no
pueden caminar y requieren una silla de
ruedas para movilizarse.
- rasgo distintivo es la capacidad de
mantener una posición sedente sin
soporte
- retrasos en los hitos del desarrollo motor
- diagnóstico casi siempre
se realiza antes de los 2
años
- dx
- AME Tipo III
- también se conoce como Enfermedad
de Kugelberg Welander, o Atrofia
Muscular Espinal Juvenil.
- DX
- Normalmente es diagnosticada
antes de los 3 años de edad, pero
es posible que no se diagnostique
hasta la adolescencia
- rasgo característico la capacidad de
ponerse de pie y caminar de forma
independiente
- dificultad para caminar, correr, y subir escaleras
a medida que se hacen mayores
- tienen riesgo de tener sobrepeso
- se puede observar un temblor leve de los dedos y las
manos, y con frecuencia se producen síntomas de
dolores y uso excesivo de las articulaciones
- dificultades para comer y dificultades respiratorias
- dificultades para comer y dificultades respiratorias
- se puede observar un temblor leve de los dedos y las
manos, y con frecuencia se producen síntomas de
dolores y uso excesivo de las articulaciones
- tienen riesgo de tener sobrepeso
- dificultad para caminar, correr, y subir escaleras
a medida que se hacen mayores
- rasgo característico la capacidad de
ponerse de pie y caminar de forma
independiente
- Normalmente es diagnosticada
antes de los 3 años de edad, pero
es posible que no se diagnostique
hasta la adolescencia
- DX
- también se conoce como Enfermedad
de Kugelberg Welander, o Atrofia
Muscular Espinal Juvenil.
- AME Tipo IV(Iniciación en la edad adulta)
- síntomas de leves a moderados
comienzan normalmente en la
segunda o tercera década de vida
- músculos utilizados para tragar y respirar
raramente se ven afectados
- impedimento motor leve paciente tiene la debilidad
muscular, temblor y las sacudidas, con o sin problemas
respiratorios
- impedimento motor leve paciente tiene la debilidad
muscular, temblor y las sacudidas, con o sin problemas
respiratorios
- músculos utilizados para tragar y respirar
raramente se ven afectados
- síntomas de leves a moderados
comienzan normalmente en la
segunda o tercera década de vida
- AME Tipo I
- tipos
- defectos en el gen SMN1, el cual fabrica una proteína que es
importante para la supervivencia de las neuronas motoras (proteína
SMN).
- niveles insuficientes de la proteína SMN llevan a la degeneración de
las neuronas motoras inferiores
- niveles insuficientes de la proteína SMN llevan a la degeneración de
las neuronas motoras inferiores
- que causan atrofia y debilidad musculares, usualmente de forma simétrica y proximal,
con predominio de extremidades inferiores y estando respetada la musculatura facial y
el intelecto
- diagnóstico
- clínicamente observando la apariencia física del niño
- pruebas genéticas
- electromiografía (EMG)
- biopsia del músculo
- biopsia del músculo
- electromiografía (EMG)
- pruebas genéticas
- clínicamente observando la apariencia física del niño
- tratamiento
- todas las terapias
- ft
- terapia acuatica
- AMA
- SDM
- Es importante que los niños
afectados con AME estén en una
posición derecha a la edad más
temprana posible. El estar de
pie es importante en el
desarrollo ya que permite una
mejor función respiratoria,
mejora la función intestinal, y
proporciona una mayor
movilidad. Es extremadamente
beneficioso que los niños sean
colocados en una posición
derecha (vertical) durante el
mayor tiempo posible, o tanto
como lo toleren, a lo largo del
día
- terapia acuatica
- ft
- LA AME no tiene aún
tratamiento curativo
- todas las terapias
- Trastorno hereditario autosómica recesiva de las neuronas motoras
espinales y bulbares .
- HEREDITARIAS
- ATROFIAS ESPINALES AME
- ADQUIRIDA
- VIRALES
- POLIOMIELITIS
- enfermedad
caracterizada por una
parálisis fláccida
asimétrica
- causada
- POLIO virus perteneciente al
género de los enterovirus,
familia Picornaviru
- transmite
- ingestión de sustancias
contaminadas
- contacto directo con personas que
puedan transmitir la enfermedad.
- ingestión de sustancias
contaminadas
- POLIO virus perteneciente al
género de los enterovirus,
familia Picornaviru
- causada
- afecta sobre todo a
los menores de 5
años
- TIPOS
- abortiva 4-8%
- Poliomielitis no paralitica
- Paralítica
- 0,5-1%Inicio es similar a forma abortiva Se recupera en 3 a 5 días (enfermedad menor) , Recae
síntomas previos con mayor intensidad, 2 a 4 días después de que principia el segundo periodo
(enfermedad mayor), se produce parálisis
- cuadro clínico
- inicio: irritabilidad, dolor muscular, manera súbita, se caracteriza por ser una lesión de las astas
anteriores de la médula, peculiaridades: a) Asimétrica , Proximal Pérdida o limitación de los
movimientos voluntarios en las zonas afectada, Flacidez o atonía , Arreflexia Sin alteraciones de la
sensibilidad, Avanza por segmentos , Afección de pares craneales: III, VII, IX, X y XI
- inicio: irritabilidad, dolor muscular, manera súbita, se caracteriza por ser una lesión de las astas
anteriores de la médula, peculiaridades: a) Asimétrica , Proximal Pérdida o limitación de los
movimientos voluntarios en las zonas afectada, Flacidez o atonía , Arreflexia Sin alteraciones de la
sensibilidad, Avanza por segmentos , Afección de pares craneales: III, VII, IX, X y XI
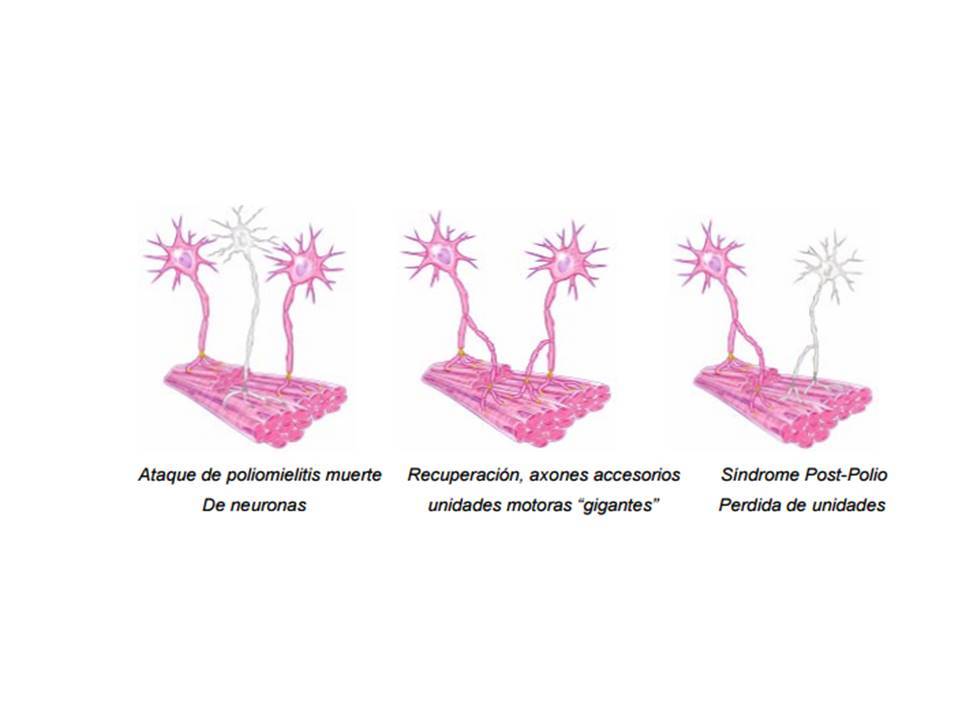
- Se estima que un 25% de los pacientes se recuperan
completamente, otro 25% queda con debilidad muscular y leves
atrofias que ocasionan escasa alteración funcional y el 50%
restante permanece definitivamente con secuelas funcionalmente
importantes
- 0,5-1%Inicio es similar a forma abortiva Se recupera en 3 a 5 días (enfermedad menor) , Recae
síntomas previos con mayor intensidad, 2 a 4 días después de que principia el segundo periodo
(enfermedad mayor), se produce parálisis
- asintomática 90-95%
- Síndrome postpoliomielitis
- se presenta entre los 15 y los 40 años después del
ataque de la poliomielitis
- sintomas
- TTO FT
- actividades de resistencia de bajo i
- aeróbicos de bajo impacto
- 3V POT SEMANA
- i :20%
- AUMENTAR mAX 10% AL MES
- AUMENTAR mAX 10% AL MES
- i :20%
- 3V POT SEMANA
- técnicas de respiración adecuada
- – Ejercicios Acuáticos
- Corrección del alineamiento postural
- Tratamiento del dolor
- Aplicación de modalidades terapéuticas
- Movilización articular
- actividades de resistencia de bajo i
- se presenta entre los 15 y los 40 años después del
ataque de la poliomielitis
- abortiva 4-8%
- consecuencias de la enfermedad
- deformidades
- estáticas
dinamicas
- Pie equinovaro supinado, pie equino, pie valgo
pronado, calcáneo talo, genu flexus, genu
recurvatum, músculos flexores de cadera
débiles
- estáticas
dinamicas
- Dificultades para la
bipedestación y la marcha
- por
- Deformidad - Debilidad
muscular - Dolor -
Control neurológico
deficitario
- Deformidad - Debilidad
muscular - Dolor -
Control neurológico
deficitario
- músculos más comprometidos en
orden de frecuencia decreciente
- tibial anterior, peroneos, tibial posterior,
extensor común de los dedos, cuádriceps,
tríceps sural, glúteos y desequilibrio de los
abductores
- tibial anterior, peroneos, tibial posterior,
extensor común de los dedos, cuádriceps,
tríceps sural, glúteos y desequilibrio de los
abductores
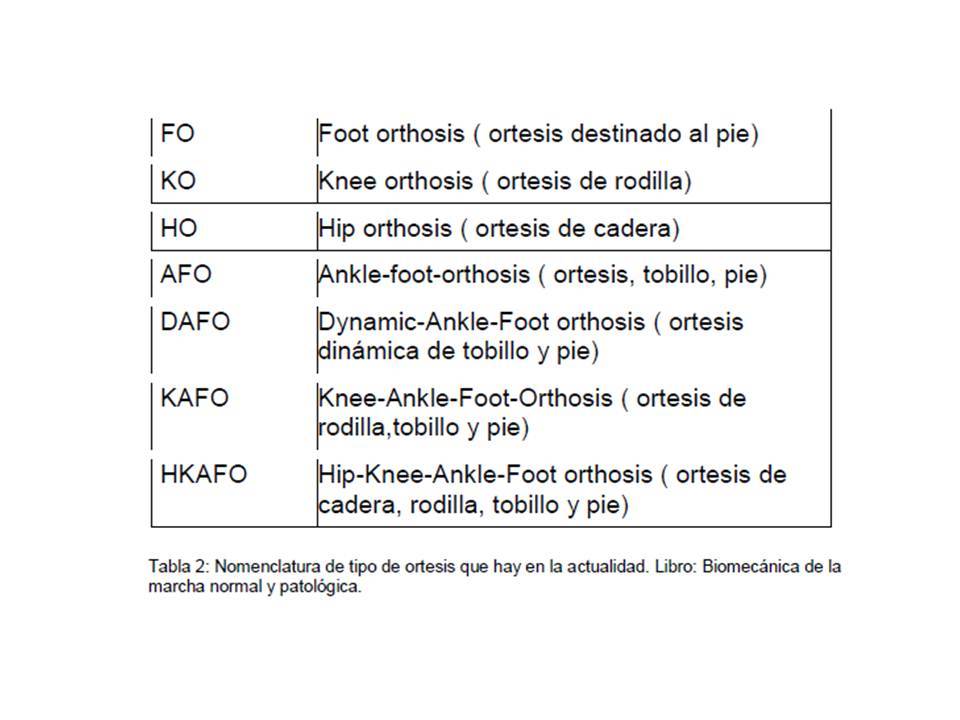
- Tto ortopedico
- A nivel del pie - A nivel de la pierna y
el pie. - Por encima de rodilla. -
Extremidad inferior y columna
- A nivel del pie - A nivel de la pierna y
el pie. - Por encima de rodilla. -
Extremidad inferior y columna
- deformidades
- MUY CONTAGIOSA
- enfermedad
caracterizada por una
parálisis fláccida
asimétrica
- fisiopatología
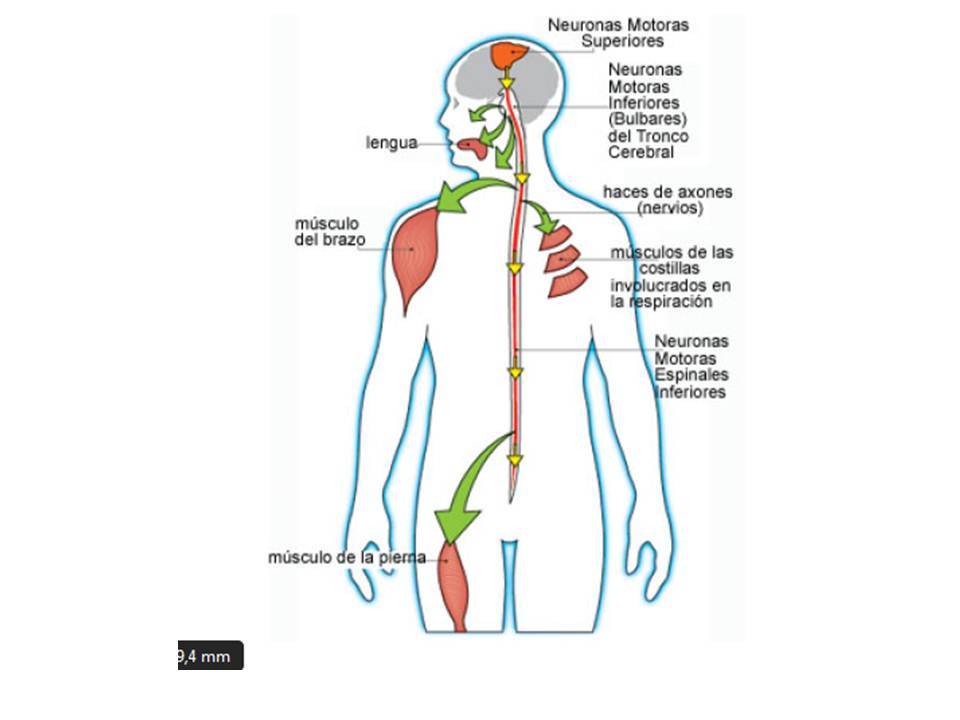
- Las regiones más afectadas del sistema nervioso central son:
- 1) Médula espinal, especialmente astas anteriores
(motoras). 2) Tronco cerebral, de este los núcleos
vestibulares y pares craneales, y el más importante la
formación reticular (aquí se encuentran los centros vitales,
respiración, ritmo cardiaco etc,). 3) Cerebelo, sobre todo el
techo y vermis. 4) Cerebro medio, materia gris. 5) Tálamo e
hipotálamo. 6) Corteza cerebral (motora).
- parálisis flácida es la expresión más evidente de la lesión de
las neuronas motoras, la atrofia muscular se debe a la
denervación, el dolor muscular y rigidez se deben a la lesión
del tronco cerebral y las arritmias cardiacas, hipertensión
arterial e insuficiencias respiratorias a la lesión de la
formación reticular (centros vitales).
- parálisis flácida es la expresión más evidente de la lesión de
las neuronas motoras, la atrofia muscular se debe a la
denervación, el dolor muscular y rigidez se deben a la lesión
del tronco cerebral y las arritmias cardiacas, hipertensión
arterial e insuficiencias respiratorias a la lesión de la
formación reticular (centros vitales).
- 1) Médula espinal, especialmente astas anteriores
(motoras). 2) Tronco cerebral, de este los núcleos
vestibulares y pares craneales, y el más importante la
formación reticular (aquí se encuentran los centros vitales,
respiración, ritmo cardiaco etc,). 3) Cerebelo, sobre todo el
techo y vermis. 4) Cerebro medio, materia gris. 5) Tálamo e
hipotálamo. 6) Corteza cerebral (motora).
- Las regiones más afectadas del sistema nervioso central son:
- Diagnóstico
- LABORATORIO LCR , * CULTIVO DEL VIRUS Muestras de heces (etapa aguda y hasta 2 o 3 meses
después), de LCR y sangre * ELECTRODIAGNOSTICO incluye: neuroconducción sensorial, motora,
electromiografía, reflejo H y onda f , 1er estudio: después de 21 días de iniciado el cuadro ,
Repetirse a los 60 días (duda diagnóstica) Deberá ser comparativo (incluidos 4 nervios periféricos)
- LABORATORIO LCR , * CULTIVO DEL VIRUS Muestras de heces (etapa aguda y hasta 2 o 3 meses
después), de LCR y sangre * ELECTRODIAGNOSTICO incluye: neuroconducción sensorial, motora,
electromiografía, reflejo H y onda f , 1er estudio: después de 21 días de iniciado el cuadro ,
Repetirse a los 60 días (duda diagnóstica) Deberá ser comparativo (incluidos 4 nervios periféricos)
- POLIOMIELITIS
- VIRALES
- ESCLEROSIS LATERAL AMIOGROFICA ELA Zapata-Zapata CH,
Franco-Dager E, Solano-Atehortúa JM, Ahunca-Velásquez LF.
Esclerosis lateral amiotrófica: actualización. Iatreia. 2016
Abr-Jun;29(2):194-205. DOI 10.17533/udea.iatreia.v29n2a08.
- caracterizada por la degeneración gradual y muerte de las neuronas motoras.
- edad de aparición 50 años pero puede aparecer en
personas mas jovenes se da mas en hombres que en
mujeres
- edad de aparición 50 años pero puede aparecer en
personas mas jovenes se da mas en hombres que en
mujeres
- DIAGNÓSTICO
- EMG, velocidad de
conducción de los nervios, RMN
- síntomas
- MNI
- Debilidad musculaR
- se debe a la muerte
progresiva de neuronas
motoras. Se manifiesta
cuando se ha perdido el 50%
de la población de neuronas
motoras
- se debe a la muerte
progresiva de neuronas
motoras. Se manifiesta
cuando se ha perdido el 50%
de la población de neuronas
motoras
- Atrofia muscular
- pérdida de fibras musculares
producida por la denervación
- pérdida de fibras musculares
producida por la denervación
- Fasciculaciones
- debidas a alteraciones de la excitabilidad
de la membrana de la neurona motora
inferior o de su axón
- debidas a alteraciones de la excitabilidad
de la membrana de la neurona motora
inferior o de su axón
- Calambres musculares
- pueden preceder a la aparición de la
debilidad muscular
- pueden preceder a la aparición de la
debilidad muscular
- Hipotonía y arreflexia
- Debilidad musculaR
- MNS
- Debilidad muscular PARESIA
- debida a la pérdida del control inhibitorio
que ejerce la vía córticoespinal sobre las
neuronas motoras inferiores que inervan los
músculos antagonistas.
- Espasticidad
- debida a la pérdida del control inhibitorio
que ejerce la vía córticoespinal sobre las
neuronas motoras inferiores que inervan los
músculos antagonistas.
- Hiperreflexia
- Reflejos patológicos
- polisinápticos
- Signo de Babinski
- Clonus
- MMSS
- Signos de
Hoffmann y
Rossolimo
- Reflejo
palmomentoniano
- Signos de
Hoffmann y
Rossolimo
- Signo de Babinski
- polisinápticos
- Labilidad emocional
- Debilidad muscular PARESIA
- MNI
- EMG, velocidad de
conducción de los nervios, RMN
- Etiologia
- mutaciones del gen que produce la
enzima del SOD1
- enzima es un antioxidante poderoso
que protege al cuerpo del daño
causado por los radicales libres
- Genética
Infecciosa
Tóxicos
Metabólicas
Metales
Autoinmune
Otros
- Genética
Infecciosa
Tóxicos
Metabólicas
Metales
Autoinmune
Otros
- enzima es un antioxidante poderoso
que protege al cuerpo del daño
causado por los radicales libres
- GLUTAMATO
- cantidades excesivas de
glutamato mata las neuronas
- cantidades excesivas de
glutamato mata las neuronas
- mutaciones del gen que produce la
enzima del SOD1
- tipos
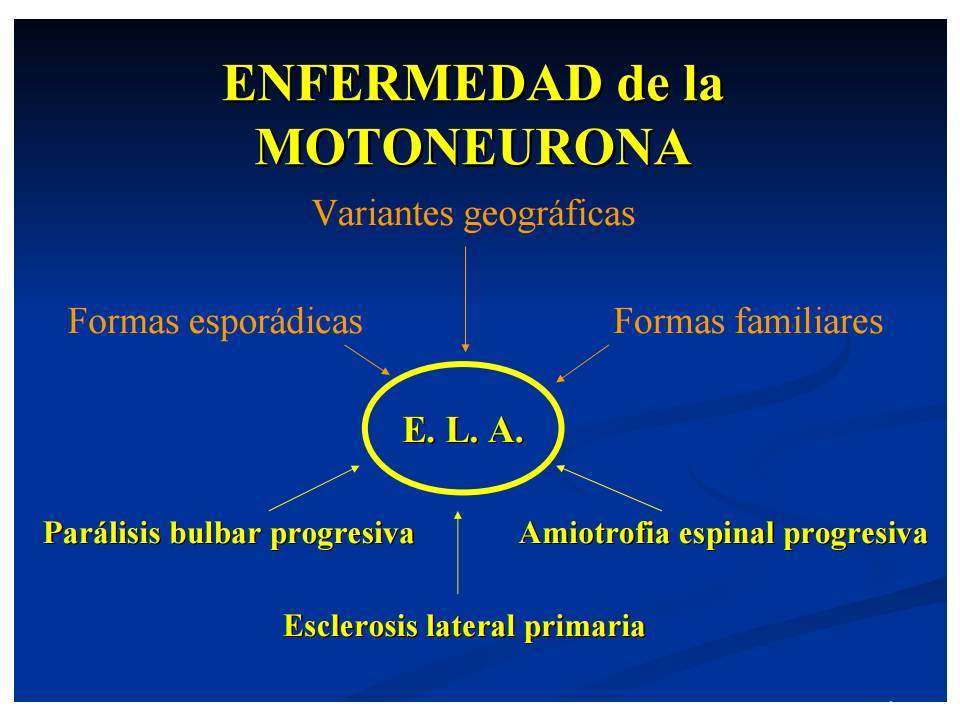
- Forma clásica de ELA
- hallazgos típicos de neurona
motora cortical e inferior ;
suele iniciarse en
extremidades superiores y
progresar en poco tiempo
hacia el resto de la
musculatura, incluida la
bulbar
- hallazgos típicos de neurona
motora cortical e inferior ;
suele iniciarse en
extremidades superiores y
progresar en poco tiempo
hacia el resto de la
musculatura, incluida la
bulbar
- Esclerosis lateral primaria
- manifiesta exclusivamente por un síndrome
de neurona motora cortical y de sus vías
córticoespinal y bulbar; la supervivencia de
los afectados suele ser superior a los 5 años
- manifiesta exclusivamente por un síndrome
de neurona motora cortical y de sus vías
córticoespinal y bulbar; la supervivencia de
los afectados suele ser superior a los 5 años
- Parálisis bulbar progresiva
- manifiesta de entrada por un síndrome bulbar (disartria, disfonía, disfagia), con
signos de neurona motora inferior bulbar (atrofia de la lengua y fasciculaciones
linguales) acompañados de labilidad emocional y signos de liberación de la vía
córticoespinal a nivel de extremidades (hiperreflexia, espasticidad).
- manifiesta de entrada por un síndrome bulbar (disartria, disfonía, disfagia), con
signos de neurona motora inferior bulbar (atrofia de la lengua y fasciculaciones
linguales) acompañados de labilidad emocional y signos de liberación de la vía
córticoespinal a nivel de extremidades (hiperreflexia, espasticidad).
- Amiotrofia espinal
progresiva (atrofia muscular
primaria)
- signos de neurona motora inferior , los signos de
neurona motora superior están ausentes. Su
progresión suele ser más lenta
- SE DEBE DIFERENCIAR DE
- amiotrofias espinales
hereditarias y neuropatías
motoras con bloqueos de
conducción.
- amiotrofias espinales
hereditarias y neuropatías
motoras con bloqueos de
conducción.
- SE DEBE DIFERENCIAR DE
- signos de neurona motora inferior , los signos de
neurona motora superior están ausentes. Su
progresión suele ser más lenta
- Forma clásica de ELA
- TTO FT
- EJERCICIO DE BAJA INTENSIDAD
- EJERCICIO AEROBICO
- ESTIRAMIENTOS
MUSCULOS NO
AFECTADOS
- atrofia muscular es severa
- MOVILIZACION ARTICULAR
- MOVILIZACION ARTICULAR
- PATRON RESPIRATORIO
- CONCEPTO BOBATH
ESPASTICIDAD
- Método Kabat (FNP).
- Masoterapia
- Ejercicios de coordinación de Frenkel.
- Hidroterapia y Natación.
- Cinesiterapia Activa Resistida
- EJERCICIO DE BAJA INTENSIDAD
- EVALUACION
- EXAMEN
MUSCULAR
- AMA
- Atrofia muscular
- Actividad refleja
- Tono
- postura
- Retracciones
- Deformidades
- marcha
- traslados y trasferencias
- patrón respiratorio
- EXAMEN
MUSCULAR
- etapas de la ela
- Etapa 1: Se sospecha ELA
- evaluaciones neurológicas completas
para excluir otras condiciones y para
hacer el diagnóstico de ELA.
- evaluaciones neurológicas completas
para excluir otras condiciones y para
hacer el diagnóstico de ELA.
- Etapa 2: diagnóstico ELA
- es necesario educar a los pacientes
y a sus familias acerca de la
enfermedad y apoyarlos al
enfrentarse a las implicaciones
- es necesario educar a los pacientes
y a sus familias acerca de la
enfermedad y apoyarlos al
enfrentarse a las implicaciones
- Etapas 3-6: ELA progresa
- Tratamientos de Apoyo.para reducir
el impacto de la enfermedad. Al
pasar por estas etapas, los
pacientes y las familias requieren
educación continua
- terapia física, ♥
terapia
ocupacional, ♥
terapia del lenguaje
♥ valoración
del estado
nutricional ♥
valoración de los
trastornos de la
respiración ♥
valoración de los
trastornos del
sueño y ♥
evaluaciones
psicológicas.
- terapia física, ♥
terapia
ocupacional, ♥
terapia del lenguaje
♥ valoración
del estado
nutricional ♥
valoración de los
trastornos de la
respiración ♥
valoración de los
trastornos del
sueño y ♥
evaluaciones
psicológicas.
- Tratamientos de Apoyo.para reducir
el impacto de la enfermedad. Al
pasar por estas etapas, los
pacientes y las familias requieren
educación continua
- Etapa 7: Enfrentándose a las decisiones del final
de la vida
- Etapa 8: La muerte, el morir y el más allá
- Etapa 1: Se sospecha ELA
- caracterizada por la degeneración gradual y muerte de las neuronas motoras.
- DEGENERATIVA
Medienanhänge
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Möchten Sie kostenlos Ihre eigenen Mindmaps mit GoConqr erstellen? Mehr erfahren.