Description

|
|
Created by Niamh McLoughlin
about 8 years ago
|
|
Page 1
Development vs discovery
Drug discovery (pre-clinical development): Lots of unknowns (including targets, binding efficiency, pharmacokinetics, toxicity etc) Difficult to plan Drug development (clinical development): Clear goals Can use information from pre-clinical phase Requires documentation & liasing with regulatory bodies
Page 2
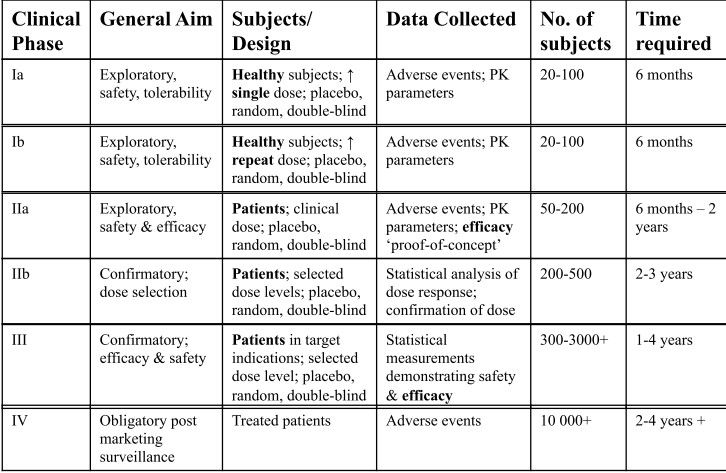
Phases of clinical development
{kind=link}
i) Regulatory requirements CLINICAL TRIALS = MOST PLACES LEGAL REQUIREMENT BEFORE NEW DRUG CAN BE SOLD/CLAIMED TO HAVE THERAPEUTIC BENEFIT/CLAIMED TO BE SAFE International regulatory requirements published by International Committee on Harmonization ICH harmonize regulation clinical trials in USA, EU & Japan - prevents unnecessary duplication of clinical research programmes in these regions Regulatory requirements for clincal trials: All trial participants must given their free & informed consent prior to participation - privacy must be assured Trial must follow a trial protocol - which has been previously approved by constituted ethics committee Only properly qualified physicians can provide medical care to trial subjects - i.e. cardiologists for cardiac drugs Clinical trial data must be recorded, handled & stored allowing accurate reporting, interpretation & verification Researchers must submit an Investigational New Drug Application - contains info regarding: Animal pharmacology & toxicology studies Manufacturing info Clinical protocols + investigator info (patient info, trial design & process, outcome measures)
Page 3
Phase I (a & b)
What happens in this stage? The drug is administered to humans for 1st time What are the aims of this phase? Establish the safety & pharmacokinetics of drug in humans Who is involved in this phase? Typically healthy volunteers
i) Phase I a) - (single dose) Aim = investigate Pk of drug &/or its metabolites after single dose of drug Small cohorts of subjects are given the drug Each cohort receives a higher dose than the last - based on either exponential or constant dose escalation Exponential = v low preclinical toxicity Constant = some preclinical toxicity Maximum tolerated dose: Highest dose than can be offered until safety & tolerability issues arise Based on 'highest dose given such in which no adverse effects were seen in most sensitive species tested in toxicology' 'No observable adverse effect level' Patients constantly monitored - heart rhythm & rate, BP, resp rate, temp, liver & renal function Blood samples taken before dosing & @ 10, 20, 30 mins, 1, 2, 4, 6, 8, 12, 24, 48 hours ** ALL ADVERSE EFFECTS RECORDED - CATEGORISED BY SEVERITY, DURATION, OUTCOME & CAUSALITY
Why are blood samples taken? Measure Pk profile: Max conc. Drug half life Mean residence time Drug clearance Area under curve Volume of distribution Food effect - Well tolerated dose given to cohort on 2 occasions - 'fed' & 'fasted' - test impact of food on Pk parameters
a) Healthy subjects & selection criteria Most volunteers young healthy males Why? Testing on females could lead to fertility issues Potentially harm foetus Sometimes specific age groups or ethnicities are required: Elderly patients for dementia drugs If a drug tested primarily on caucasians approved in Asia Cancer patients often used in Phase I of cancer drugs Due to inherent drug toxicity - unethical to test on healthy, cancer-free patients
b) Eligibility criteria Informed consent Patient understands risks, benefits, procedures, renumeration, insurance & their rights Full physical exam Medical history ECG Vital signs (BP, heart & resp rate, temp) BMI Blood & urine tests Serology for viral infections (Hep & HIV) Alcohol breath test Nicotine test Blood sample Pk & safety Exclusion criteria Rules person out of participating in trial High BMI Cardiovascular abnormalities Drug hypersensitivites Recent clinical trials Blood donations
c) Ethics All human studies performed to strict ethical requirements, as set by Declaration of Helsinki & Nuremberg codex http://www.nejm.org/doi/full/10.1056/NEJM199711133372006#t=article Belmont report = basic ethics to be adopted in human experimentation 3 principles: Respect for persons - treat individuals as autonomous agents Beneficence - maximise possible trial benefits & minimise harm Fairness in distribution - equal opportunity to participate in trials 3 recommendations: Avoid bias Publication of findings Informed patient consent
iii) Phase I b (repeated-dose) Aim = Investigate whether Pk of drug &/or its metabolites change after repeated dosing Want to investigate drug accumulation: Is the drug causing saturation of pathways? Is the drug inhibiting drug-metabolising enzymes? If yes - could lead to toxic drug build-up! Also want to investigate induction drug metabolising enzymes: Could lead drug conc. to drop to subtherapeutic levels! Blood samples taken at similar interval to single dose as far as 12hr for day 1 - after that 12 hr on days 2-7 & again as single-dose on day 8
Page 4
Phase II (a & b)
i) Phase II a Aim = Give evidence of safety & efficacy of drug Small group of diseased patients PK profiles often changed in disease states Researchers want to clarify: Dose regimen - Based on PK profiles from phase I + drug's MOA Duration - Depends on indication (Efficacy for post-operative pain can be shown quickly vs months/years for Alzheimer's) In order for the drug to advance to phase II b) (Proof of concept (POC)) - drug must fufill the following criteria: If it is a First in class drug, the POC must be for the entire new class of drugs - not just the specific drug in question (i.e. the mechanism of action is efficacious) If it is a Follower drug, the POC must show it is at least as efficacious as current drugs of its kind + show an improvement
ii) Phase II b Aim = Confirm efficacy & optimal dose & regimen Larger group of diseased patients - greater statistical weight due to increased group number In this stage - explore the dose-response relationship A dose for large-scale phase III studies is selected Phase II trials have 4 design types: Parallel group Randomized Double-blind Placebo-controlled Open label: Cancer chemotherapy - unethical to use placebo Want to find primary outcome measure - certain change from baseline response must occur
iii) Trial design 'Between-patient comparison': Most trials do this 'Within-patient comparison': Part of cross-over trials Individuals get more than one treatment Inter-patient variability minimized Short term - cannot study curative medicines 'Multicentre': Trial run in more than one location/country/continent Patient accural faster Conclusions more representative of population Complex planning involved More expensive Requires standardised training Problems faced in trial design include: Appropriate evaluation period Carry-over effects (Wash-out period where participants discontinue treatments they were using before clinical trial) Run-in period Period before clinical trial commenced Can be passive or active (Patient wasn't or was having treatment) Randomization
iv) Randomisation & blinding mechanisms During trials - important to avoid BIAS Randomisation = Method of assigning trial participants to treatment groups 3 types of blinding mechanisms: Single blinding: Patient does not know what treatment they receive Double blinding: Neither patient nor clinician knows which treatment the patient is receiving Triple blinding: Neither patient, clinician nor statistician knows what treatment patient is receiving These processes: Eliminate bias Ensure significant results are real Ensure each group is average & equal with similar characteristics
Page 5
Phase III
Aim = Measure efficacy, safety & Pk in disease state Large scale studies in disease patients Placebo-controlled Researchers may be required to demonstrate quantifiable advantages over standard treatments Such advantages might include: Safety Tolerability Efficacy Compliance Cost-effectiveness Will normally compromise several studies - multicentre (regions where drug is sold) Intrinsic (genetic) & extrinsic (diet) differences affecting safety & efficacy need bridging studies: Supplemental study performed in new region to provide PK or clinical data on efficacy, safety, dosage & dose regimen in new region that will allow extrapolation of foreign clinical data to new region Must be carried out due to polymorphisms in cytochrome P450 enzymes in different populations
Want to create your own Notes for free with GoConqr? Learn more.